Alzheimer’s disease is one of the deadliest and costliest diseases on Earth. It kills hundreds of thousands of Americans every year, afflicts an estimated 11% of all Americans 65 and older in 2026 (roughly 7.4 million people), and costs Americans $340 billion annually. In 2024, it was the fifth-leading cause of death among people over 65. And it’s only getting worse. Total deaths from Alzheimer’s in the US increased more than 3x from 2000 to 2021. Globally, annual deaths from Alzheimer’s have grown from 621.7K in 2000 to over 1.8 million in 2021.
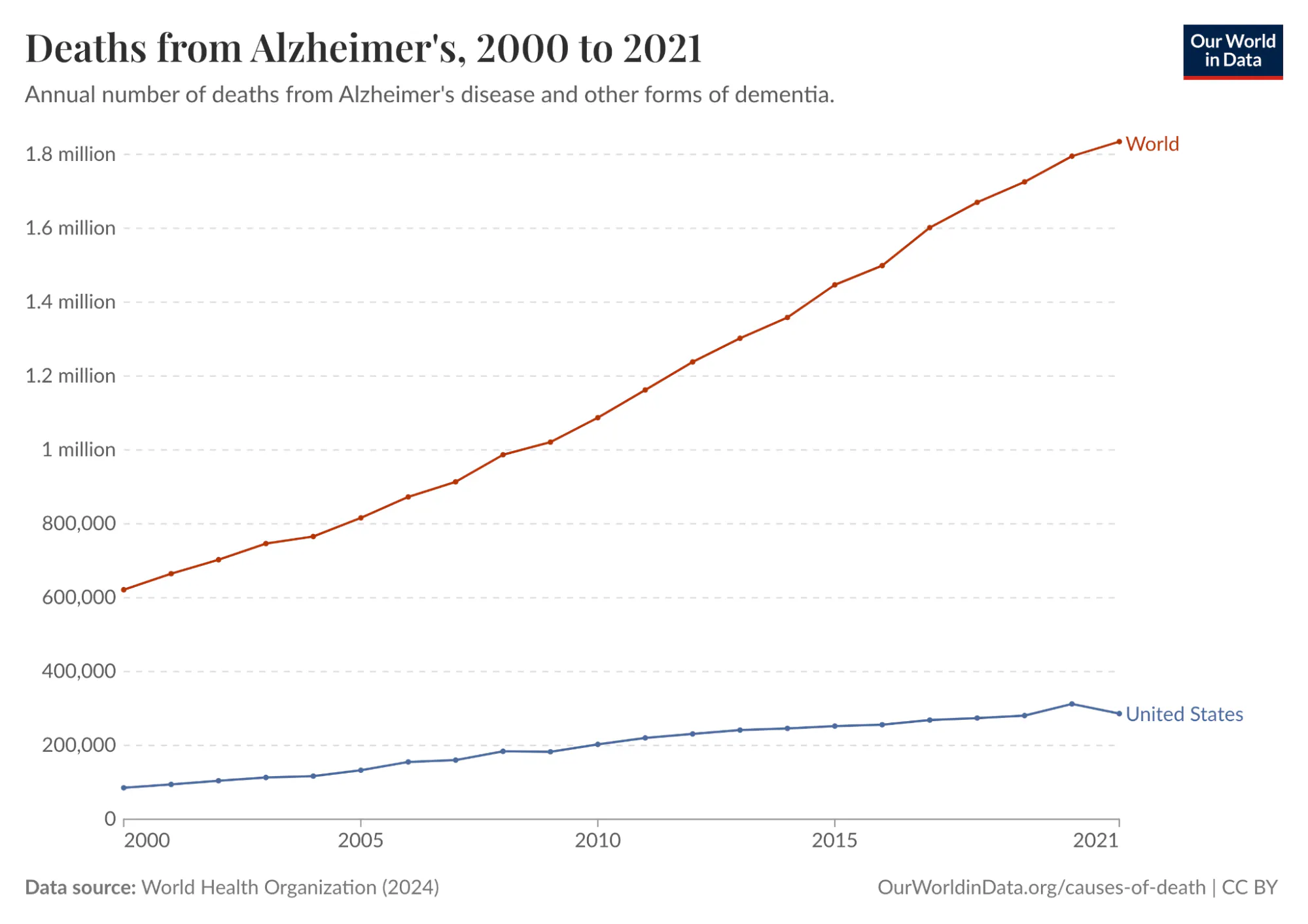
Source: Our World In Data
Despite all this, there has been no meaningful progress towards a cure, even though enormous resources have gone into Alzheimer’s R&D. There was an estimated $42.5 billion of private investment in Alzheimer’s R&D between 1995 and 2021 alone, but much of this investment was a complete waste. One study conducted in 2014 found a 99.6% failure rate in Alzheimer’s drug clinical trials conducted between 2002 and 2012. As a result, the FDA did not approve a single new Alzheimer’s treatment in the 18 years between 2003 and 2021.
A big reason for the wastefulness of Alzheimer’s research was that much of it relied on an imperfect proxy as its starting principle. For over two decades, the leading hypothesis for Alzheimer’s was the "amyloid cascade": the idea that toxic protein clumps called amyloid-beta plaques were the primary driver of the disease. Almost every major pharmaceutical company based its Alzheimer’s drug development on this idea.
The problem was that the mouse models used to validate it were engineered to develop amyloid plaques, but they never actually had Alzheimer's. They lacked the tau protein tangles, the widespread neuronal death, and the microglial dysfunction that research suggests is central to how the disease progresses in humans. When drugs cleared plaques in mice and appeared to restore cognition, they succeeded in treating the model. But when those drugs entered human trials, despite successfully clearing plaques in patients as predicted, they had little to no effect on cognitive decline.
Ultimately, Alzheimer’s drugs had such a high failure rate, both because they went after the wrong target and because they used mouse models as an inaccurate proxy to test a treatment meant for human beings. Mouse models don't develop Alzheimer's the way humans do, although they were convincing enough to sustain a $42.5 billion research program for 25 years at a 99.6% failure rate. In this way, Alzheimer’s exemplifies the underlying problem with the status quo in drug development: we rely on imperfect biological proxies until we get human trial data, limiting the speed and efficacy of drug development at enormous cost in time, money, and lives.
Relying on such proxies affects all three stages of drug development. The first stage is targeting: identifying which molecule in a cell is causally responsible for a disease. The second is treatment: designing a drug that reaches that target without causing unacceptable side effects elsewhere. The third is trials: testing the drug in the right patients, at the right dose, measuring the right outcomes. Failure at any of these stages can lead to years of wasted work and enormous financial loss.
The inherent inefficiencies of this paradigm have led to a phenomenon known as Eroom’s Law, which holds that drug discovery has become exponentially slower and more expensive since the 1980s. Despite dramatic advances in molecular biology, genomics, and screening technologies, the cost of bringing a single new drug to market has risen to over $2 billion, while 90% of drugs that enter clinical trials fail.
Clearly, something needs to change. A new paradigm is needed. The most promising candidate for that shift is the virtual cell: a computational model trained on human biological data that simulates how a cell responds to a drug or genetic change. Instead of inferring human biology from a mouse or other animal's drug response, a virtual cell learns the rules of human cellular behavior directly from human data, including which genes are active, how they regulate one another, and how that network shifts under disease or intervention.
In the context of Alzheimer's, a well-trained virtual cell might have revealed earlier that amyloid plaques were a symptom of a deeper dysfunction, driven partly by the brain's immune cells becoming locked in a self-reinforcing inflammatory state, rather than the root cause. It would not have been a guarantee, but it could have narrowed the target list faster and flagged the mismatch between mouse and human biology sooner.
This essay is intended to paint a picture of virtual cells and their potential to help finally overthrow the long stagnation in drug development described by Eroom’s Law. It begins by diagnosing why drug development is so slow and costly, tracing the history of biological proxies from the first animal testing requirements in 1938 to the organ-on-chip systems the FDA is validating today. It then describes what virtual cells are and why now is the right moment to build them. From there, it surveys the diseases for which virtual cells are most likely to make a difference. Finally, it lays out the key companies in the virtual cell landscape and describes how virtual cells may usher in a new era for drug development.
Of the many promising potential applications of virtual cells, one can already be put in practice: using virtual cells to understand how the same diseases present differently across a patient population, i.e. for patient stratification. In the future, virtual cells may well become the foundation of a faster, more accurate, and lower-cost testing regime, and thereby bring about a new renaissance in drug discovery. However, existing testing techniques, such as animal testing, will be unlikely to disappear entirely, as they will likely continue to be useful supplements to virtual cells even if their full potential is realized.
Why Drug Development Is Broken
Eroom’s Law is Moore’s Law spelled backward because it represents a phenomenon that is the mirror image of Moore’s Law, a decades-long trend in which computing power became both exponentially more powerful and cheaper at the same time. By contrast, over the past 70 years, the number of new drugs approved per dollar of R&D spending has roughly halved every decade.
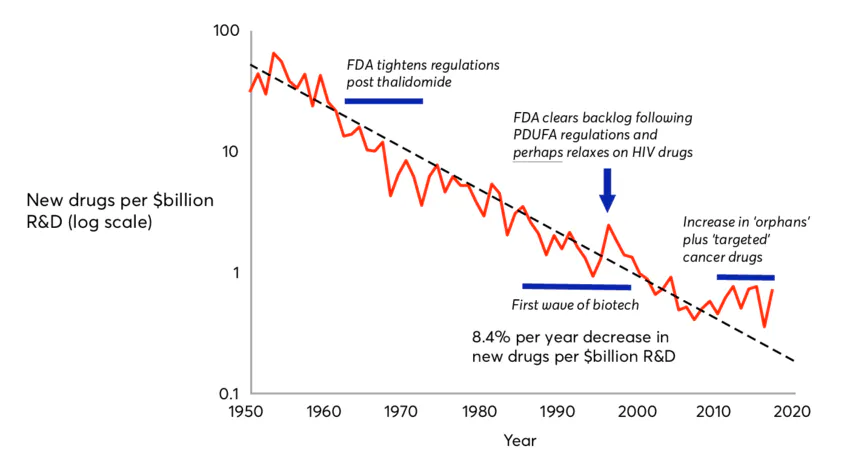
Source: Wikipedia
The advent of AI pits Moore’s Law against Eroom’s Law. The virtual cell is an application of AI in biology that has the potential to translate progress in computing into advances in medical treatment, and thereby fulfill the lofty predictions of frontier AI labs. It bridges the gap between the world of AI and the world of cells. But to understand the potential of virtual cells going forward, it is important to understand the limitations of the drug discovery process as it functions today. As mentioned earlier, there are three broad reasons why 90% of drugs fail to reach patients today; three possible points of failure in the drug discovery pipeline: targeting, treatment, and trials.
Failure Mode 1: Targeting
To cure a disease, you first need to know what's causing it. There are typically many molecules in a cell, such as DNA, RNA, and proteins, that could cause disease. A single human cell contains an estimated several billion protein molecules, and identifying the right target for a drug is difficult. The Alzheimer's story illustrates what happens when the wrong target is chosen: billions and decades spent pursuing amyloid plaques, a target that turned out to be a symptom rather than a cause.
But the amyloid case is not unique. A single diseased cell can contain hundreds of plausible molecular targets, and without a way to systematically narrow that list, picking the right one is largely a matter of trial and error. This is almost certainly one of the forces compounding Eroom's Law: the more potential targets we discover through advances in genomics, the harder it gets to identify the right one.
Failure Mode 2: Treatment
Even when the right target is chosen, a drug can still fail if it can't reach that target effectively, or if it causes unacceptable side effects in the process. Pfizer's torcetrapib is a stark example. The drug was designed to raise HDL, or "good" cholesterol, as a treatment for cardiovascular disease, and it worked: HDL levels rose by 72% in trials. Pfizer's CEO called it "one of the most important compounds of our generation."
Three days later, Pfizer halted its Phase 3 trial after the drug showed a 58% higher rate of death compared to placebo. It turned out the drug was stimulating hormone secretion from the adrenal glands, a side effect entirely unrelated to its intended target. Pfizer had invested approximately $800 million in the drug, and its stock fell 10% on the day the trial was halted, wiping out $21 billion in market cap and clearly illustrating how costly failures can be when treatments have unintended effects that are nearly impossible to predict.
Failure Mode 3: Trials
Even with the right target and a drug that reaches it safely, a clinical trial can still fail if it enrolls the wrong patients. AstraZeneca's MYSTIC trial in 2017 is one example. The trial tested a combination of two immunotherapy drugs against non-small cell lung cancer in an effort to compete with Merck's Keytruda. By 2017, the field had already established that response to these drugs depended heavily on the level of PD-L1 expression in a patient's tumor.
Merck had set its patient selection threshold at 50% PD-L1 expression, a cutoff that enriched its trial for likely responders and helped make Keytruda the world's best-selling drug. AstraZeneca, by contrast, set its threshold at 25%, diluting its trial population with patients unlikely to respond. The MYSTIC trial failed its primary endpoint, and AstraZeneca's stock fell 16% in a single day, wiping out about $10 billion in market cap. The difference between thresholds of 25% and 50% costs a decade of progress for patients and billions for shareholders, exemplifying the broader principle that clinical trial design is critically important to drug development but very difficult to do based on the educated guesses that drug developers must rely on based on our limited understanding of the underlying biology.
Our Limited Understanding of Biology
These three failure modes share a common root. In each case, the underlying problem is an insufficient understanding of human biology at the cellular level. The wrong target was chosen in Alzheimer's because mouse models gave a misleading signal. Torcetrapib's fatal side effect went undetected because no preclinical model predicted what the drug would do to human adrenal hormone secretion. AstraZeneca set the wrong patient-selection threshold because PD-L1 expression is a crude proxy for the far more complex cellular dynamics that determine whether a patient's immune system will respond.
In each case, an imperfect biological proxy produced a plausible but ultimately false signal, hundreds of millions of dollars and years of work followed, and human trial data eventually made it clear that the wrong path was being pursued. Understanding why this keeps happening requires a closer look at the tools we use to model human biology and why they’re so limited.
The Preclinical Testing Problem
The modern framework for drug testing was born from tragedy. In 1937, a pharmaceutical company released a new antibiotic dissolved in a solvent called diethylene glycol without any safety testing. The resulting drug killed 107 people, many of them children, who died from acute kidney failure. The public outcry forced the passage of the 1938 Federal Food, Drug, and Cosmetic Act, which for the first time required manufacturers to demonstrate drug safety before marketing. In practice, this meant animal testing became the de facto safety standard for the next few decades.
A second watershed moment came in the early 1960s, when thalidomide, a sedative marketed in Europe as safe for pregnant women, caused an estimated 10K to 20K cases of severe birth defects worldwide. The 1962 Kefauver-Harris Amendments that followed codified animal testing as a legal requirement for drug approval in the United States. That requirement stood, largely unchanged, for the next 60 years. The FDA Modernization Act 2.0, passed in 2022, was the first legislation to formally permit alternatives to animal testing in preclinical drug development.
Today, preclinical drug testing relies on five primary models, each representing an incremental attempt to better approximate human biology: mouse and rat testing, two-dimensional cell cultures, non-human primate testing, three-dimensional cell cultures, and organ-on-chip systems. Beyond this, there is the newer approach of ex vivo whole-organ perfusion. Each of these preclinical testing models has flaws that contribute to the high failure rate of drugs.
Mouse and Rat Testing

Source: Charles River Laboratories
Mouse and rat models remain the most widely used in drug testing, accounting for the vast majority of laboratory animals in biomedical research. This is because they’re cheap, breed quickly, and their genomes were among the first to be fully sequenced, making them easy to modify genetically.
Since 1980, researchers have inserted human genes into mice to better simulate human drug responses. These "humanized" mice are a better proxy than natural mice, but fundamental problems remain stemming from biological differences between mice and humans. For instance, human blood is dominated by neutrophils, the immune system's first responders, making up 50 to 70% of circulating white blood cells. In mouse blood, 75 to 90% are lymphocytes instead.
The two species don’t just differ in their immune systems; the liver enzymes that metabolize drugs also differ substantially between species, meaning a drug safely cleared by a mouse may be converted into a toxic compound in a human, or vice versa. And mice live two to three years, meaning diseases that unfold over decades in humans must be artificially compressed, distorting the disease progression in ways that are difficult to account for.
Two-Dimensional Cell Cultures
Two-dimensional cell cultures, in which cells are grown on flat plastic surfaces, have existed alongside animal models since the 1950s and remain the dominant tool for high-throughput screening of drug candidates. Their limitations are equally well established. Cells in living tissue exist in three-dimensional environments with complex structural and chemical interactions that regulate gene expression and drug response. In 2D, cells flatten and behave in ways that diverge substantially from their behavior in the body. One study found that cancer cells in 3D cultures are 2-5x more resistant to chemotherapy than the same cells in 2D, suggesting that 2D results may overestimate drug efficacy.
Three-Dimensional Cell Cultures
Three-dimensional cell cultures, including stem cell-derived organoids, have become increasingly standard in oncology drug development, toxicity screening, and disease modeling. While organoids restore tissue architecture, they typically lack blood vessels, limiting oxygen and nutrient delivery needed to simulate human biology, and also lack immune cells, which are critical for testing drug safety and efficacy. Organoids are also more expensive and variable than 2D cultures, limiting their use in large-scale screening.
Non-Human Primate Testing
Source: Fortune
Non-human primates are used when rodent models are deemed insufficient, particularly for antibody and vaccine development. Despite sharing closer genetic identity with humans than rodents, they still fail to reliably predict human drug responses. A 2015 study of over 3K drugs found that the absence of toxicity in animals provides little evidence that adverse reactions will also be absent in humans, even when the animal data comes from primates. The financial and ethical barriers of this type of testing are also extremely high: testing a single drug in a non-human primate can cost as much as a human clinical trial.
Organ-on-Chip Systems
Organ-on-chip systems are microfluidic platforms lined with human cells and perfused with physiological fluids that can replicate drug absorption, metabolism, and excretion in a single platform. A 2022 study found these systems modeled liver toxicity with higher accuracy than animal models. The FDA is formally developing validation frameworks for their use under the New Approach Methodology framework introduced by the 2022 Modernization Act. Although they show promise, organ-on-chip systems can’t yet fully replicate neuroendocrine signaling, circadian rhythms, or long-term aging effects, and formal regulatory standards for their use are still being written.
Ex Vivo Whole-Organ Perfusion
This is a newer approach that bypasses the problem of modeling human biology entirely by testing drugs directly on intact human organs. In ex vivo whole-organ perfusion, organs donated from deceased individuals are connected to machines that circulate oxygenated, nutrient-rich fluid through the organ's blood vessels, maintaining cellular activity outside the body for up to 24 hours. The foundational breakthrough came in 2019, when Yale neuroscientists published a paper demonstrating that molecular and cellular functions could be partially restored in pig brains hours after death. Their startup, Bexorg, now applies this technique to donated human brains to test drug candidates for Alzheimer's and Parkinson's in intact human tissue before clinical trials.
Beyond the brain, organizations such as TNO in the Netherlands have applied the same approach to human livers and kidneys in drug metabolism studies. It is the closest any preclinical system can get to testing drugs in living human tissue. But it is inherently constrained by donor organ supply, limited perfusion windows of only a few hours, and the inability to observe chronic or systemic effects, making it a powerful but narrow tool for specific pharmacological questions rather than a scalable platform for drug discovery.
A New Preclinical Testing Paradigm Is Badly Needed
After 90 years of mandated animal testing, the scientific evidence is unambiguous. Animal and in vitro models have a high rate of translational failure, exceeding 90% in most analyses, primarily due to unexpected toxicity or lack of efficacy in humans. Species differences are fundamental and cannot be controlled for because they reflect millions of years of divergent evolution.
As a result, regulatory frameworks are finally shifting towards more human-relevant methodologies, with the FDA explicitly phasing out animal-testing requirements for antibodies. The FDA Modernization Act 2.0 and the 2025 FDA Roadmap explicitly recognize that the future of preclinical testing lies in New Approach Methodologies encompassing organ-on-chips, organoids, computational models, AI/ML-based toxicity prediction, and patient-derived in vitro cells.
The need for a paradigm shift in preclinical testing is paramount. While test-tube cells, mouse studies, and primate studies have served a purpose, they are no longer adequate proxies for complex human diseases. We cannot afford to waste millions of dollars and years on R&D before getting our first human data samples back. But testing drugs early in humans is too risky until we’ve done enough drug safety research.
Computational tools offer a solution to this problem. We could try to simulate the effect of a drug on a diseased virtual human. If we could collect enough data on real human biology, we could train AI models to understand biological rules that we might be decades away from discovering through trial and error. But before we can simulate the human body as a whole, we must first simulate its parts. Starting with individual cells, then the tissues they form, then the organs those tissues build, and only then the full organism.
Virtual Cells: A Potential Paradigm Shift
A virtual cell is a computational model of a cell that simulates its molecular mechanisms and behavior, and can therefore predict how a cell will respond to a drug or genetic change without conducting a physical experiment.
Although the term “virtual cell” is relatively recent, the concept has a long history in systems biology. One of the most prominent early efforts came from the Covert Lab at Stanford, which in 2012 published a whole-cell model of Mycoplasma genitalium, a simple bacterium with a small genome. The team mapped every gene by hand and built equations capable of predicting the bacterium's phenotype from its genetic sequence alone.
This was the first demonstration that a computational simulation of a living cell was technically achievable. More recent work has pushed further: a 2026 study simulated the entire life cycle of a genetically minimal bacterium, tracking every molecule's position inside the cell as it grew, replicated its DNA, and divided, with the virtual cell's predicted division time falling within two minutes of the real bacterium's 105-minute cycle. The study demonstrated that with enough data and computation, it’s possible to recreate cellular behavior inside a computer.
However, as cell modeling research moves on from whole-cell bacteria models to more complex organisms, it is important to note that these early efforts succeeded by keeping the scope narrow: small genomes, exhaustive manual curation, and carefully enumerated molecular processes. Those constraints made it possible to create a complete mechanistic model. That approach works for simple bacteria, but not for more complex human cells, which contain roughly 20K genes and have hundreds of distinct cell types.
That’s where first-generation efforts differ from modern-day virtual cells. While early efforts relied on building algorithms based on our understanding of biology, current methods show AI models how cells behave empirically, and the models learn the underlying biology. This approach aligns with “The Bitter Lesson”, an AI principle first articulated in a 2019 blog post that observes that over the long run, AI models that scale with computation, thanks to Moore’s law, consistently outperform hand-crafted domain insight.
Applied to virtual cells, the bitter lesson implies that AI models should not be limited by human understanding of biology, but instead by the richness of the data and computation they have access to. That said, the bitter lesson only comes into effect once data and computation cross a critical threshold. Until mid-2025, virtual cell models had not yet reached that point, and most could not outperform simple statistical baselines like linear regression.
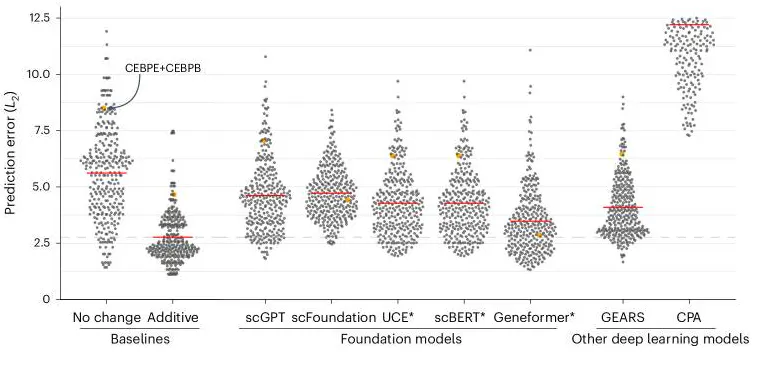
Source: Nature Methods
However, that has now changed because of two converging revolutions. Together, these revolutions have created conditions under which building a virtual cell has become a serious scientific goal rather than a thought experiment.
The first is the exponential collapse in sequencing costs and subsequent rise in open-source datasets. In 2001, sequencing a single human genome cost approximately $100 million. Today it costs around $100. This cost collapse has unlocked the ability to profile the molecular state of individual cells at massive scale. Because DNA encodes RNA, and RNA encodes proteins, cheaper DNA sequencing has directly enabled cheaper RNA sequencing, which reads out which genes are active in a cell at a given moment, providing a snapshot of the cell's functional state.
The practical consequence is profound. For the first time, we have the scale of raw material to train AI models on human biology at cellular resolution. A virtual cell model requires large amounts of data to learn the rules governing how a cell responds to changes or perturbations, such as a drug treatment or knocking out a gene.
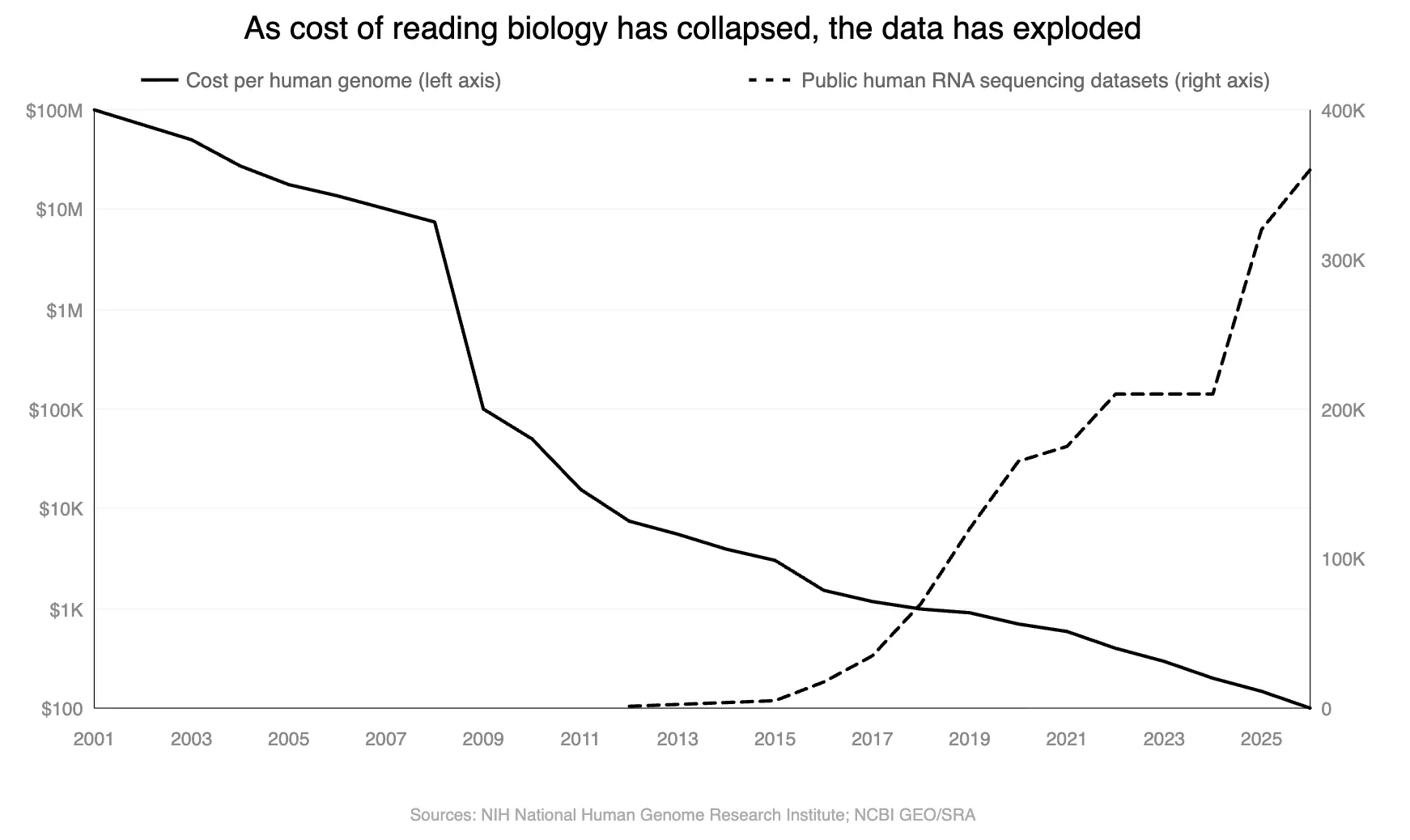
The second revolution is the maturation of AI architectures capable of learning complex biological rules from data at a scale no human scientist can match. In 2020, researchers at OpenAI documented that AI models improved as a power law function of compute, data, and model size, a finding subsequently observed across vision, language, and reasoning tasks.
Whether scaling laws hold for biology is a question with partial but encouraging answers. In protein modeling, they do: the ESM family of protein language models showed systematic improvements in structure and function prediction as model size increased, with atomic-resolution structural information emerging in learned representations as models scaled to 15 billion parameters. Earlier work on ESM-1b demonstrated that biological structure and function emerge from unsupervised learning on protein sequences alone.
Biology does not, however, satisfy all the conditions that make scaling work cleanly in other domains. Internet text is abundant, cheap, and information-dense. Biological data is sparse, heterogeneous, expensive, and confounded by non-biological factors. Raw data volume is not the bottleneck: the NIH DNA sequence archive exceeds 47 petabytes. But human genomes are 99.9% identical, measurements are noisy, and the signals that matter most (disease-causing variants, drug responses) are rare. More data does not necessarily mean better models, even for modalities and assays beyond DNA sequencing.
AlphaFold2 illustrates the point precisely: it succeeded not through brute-force scale but through domain-specific architecture trained on the Protein Data Bank, a dataset curated at atomic resolution by thousands of PhD scientists over decades. The lesson is that AI discovers biological rules when the data is high-quality, the objective is well-defined, and the architecture fits the problem. The question for virtual cells is whether all three conditions can be met simultaneously.
There is reason to believe they can. Over the past decade, a new category of biological experiments has emerged, changing what AI models can learn from cellular data. In 2016, researchers combined gene editing through tools like CRISPR with single-cell RNA sequencing to create Perturb-seq: disable a specific gene inside a cell using CRISPR, then measure how all ~20K genes in that cell respond. This data is not observational. It is experimental cause and effect at cellular resolution.
The 2016 experiments perturbed dozens to hundreds of genes. In 2022, research scaled this approach to perturb nearly every expressed gene in the human genome in a single experiment, generating 2.5 million single-cell readouts. As of 2025, there were efforts to generate datasets with over 1 billion cells.
This is what has changed from two or three years ago. The cost collapse in single-cell sequencing has produced the first perturbation datasets with both the scale and the interventional structure needed to train models that learn cause and effect rather than correlation. The training objective these datasets enable is concrete: given a genetic perturbation, predict the resulting shift in gene expression across the cell.
Unlike designing a “better” drug, this can be evaluated by holding out known perturbations and comparing predictions to experimental measurements. The central open question is generalization: can models trained on observed perturbations predict the effects of perturbations they have never seen in cell types they were not trained on? That is where the value lies for drug discovery.
What Is a Virtual Cell?
In 2024, researchers from Stanford, Genentech, the Arc Institute, and the Chan Zuckerberg Initiative published a collaborative paper laying out a shared vision for what a virtual cell should be:
"Our view of a virtual cell is a learned simulator of cells and cellular systems under varying conditions and changing contexts, such as differentiation states, perturbations, disease states, stochastic fluctuations, and environmental conditions. A virtual cell should integrate broad knowledge across cell biology, work across biological scales, over time, and across data modalities, and should help reveal the programming language of cellular systems and provide an interface to use it for engineering purposes."
But before looking further at what virtual cells could become, it is important to be clear about what they are not today. No existing model can fully capture all cellular processes, including DNA transcription, RNA translation, metabolism, cell signaling, spatial organization, and temporal dynamics, at a faithful resolution. Current virtual cell models do not generalize reliably across all cell types, organisms, or perturbations. Most claimed model performance is limited to particular cell lines, developmental stages, or experimental conditions. A virtual cell is, therefore, at present, not a substitute for in vivo biology. It is a tool for searching a larger space of experimental possibilities and guiding hypothesis generation more efficiently than traditional methods allow.
What a virtual cell can do rests on a key insight about the nature of cells themselves. A human cell contains an estimated several billion protein molecules, and in principle, those molecules could combine in an astronomical number of ways. But in practice, biology's internal rules, including feedback loops, regulatory dependencies, and evolutionary constraints, massively restrict which combinations actually occur. The result is that a cell's functional state, despite its underlying molecular complexity, is highly compressible. Measurements like gene expression, which captures which genes are turned on or off, and epigenomic state, which captures which genes are accessible or locked away, preserve a large share of the information needed to predict how a cell will respond to a perturbation. A virtual cell operates at exactly this level of abstraction: it learns a compressed representation of the cell that retains what is causally relevant while discarding molecular detail that is not.
The most useful metaphor for understanding how cells behave, and why virtual cells are possible, comes from the developmental biologist Conrad Waddington, who in 1957 proposed the concept of the epigenetic landscape. Imagine a terrain of hills and valleys. A cell's state is like a ball rolling across that terrain, tending to settle into stable valleys held in place by self-reinforcing gene regulatory programs. A healthy immune cell, a cancerous tumor cell, and a fibroblast depositing scar tissue each occupy their own valley. Small perturbations, like nudging the ball partway up a valley wall, won't change the cell's identity; it rolls back to where it was. But the right combination of perturbations can push the cell over a hill and into a different valley entirely, shifting it from one stable state to another.
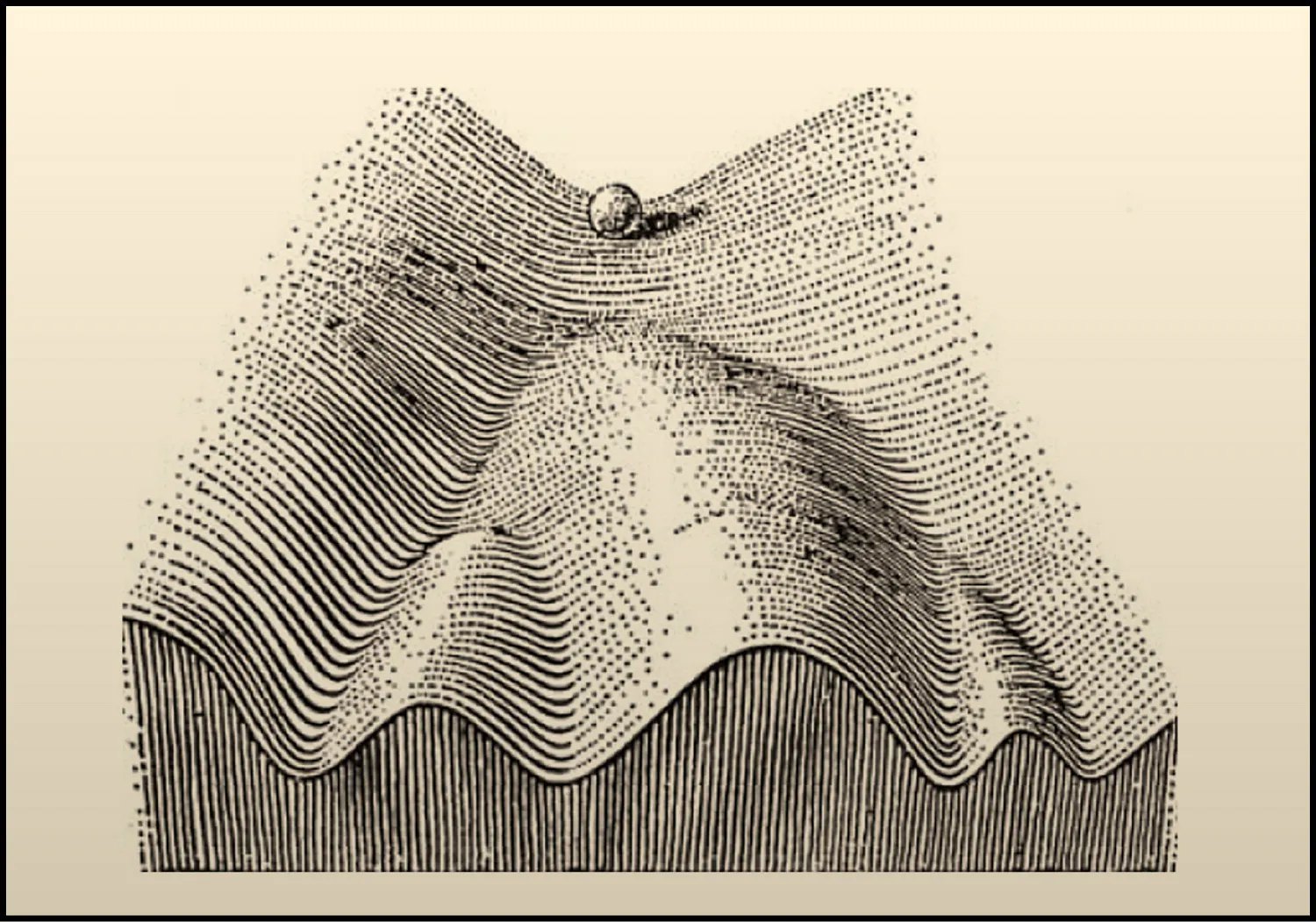
Source: Cell
Many diseases are analogous to cells getting stuck in the wrong valley, locked in a pathological state by self-reinforcing regulatory feedback. And many therapeutic opportunities correspond to finding the right push to move a diseased cell over the hill and into a healthy one. A virtual cell is, at its core, a learned map of that landscape: a model that has internalized the hills, valleys, and pathways of cellular state space well enough to predict where a cell will go when it is perturbed, and to work backward from a desired destination to identify what perturbation will get it there. By operating at the level of compressed, functional state rather than microscopic detail, virtual cells provide a principled representation of the fundamental unit of biology – one that is both computationally tractable and biologically predictive.
What Virtual Cells Can Do
A virtual cell's core capability is prediction. Given a cell's current molecular state, it can predict the cell’s phenotypic representation and how that state changes in response to an intervention, i.e., a drug or a gene knockout. But the value of that capability depends on whether the biological training data is rich enough for a model to generalize reliably. Five applications follow directly from this, both clinical and preclinical applications
Preclinical Applications
The most immediate preclinical application is forward perturbation prediction. Given a drug treatment or genetic change, a virtual cell predicts the resulting shift in the cell's gene expression profile. This enables screening of compounds and targets at a scale far beyond what is experimentally feasible. Rather than running thousands of physical experiments to test candidate drugs, a virtual cell can simulate them computationally, flagging the most promising candidates for physical validation. Because the model has learned the regulatory structure underlying perturbation responses, it can generalize to interventions it has never seen, predicting effects through pathways it has already internalized.
Toxicity prediction is the same capability as forward perturbation prediction, but rather than predicting what the drug does to the target cell, it predicts what the drug does to liver cells, heart cells, or other off-target cells. This is a high-value application: roughly 30% of clinical trial failures are attributed to unexpected toxicity. A virtual cell trained on healthy tissue cell states can predict whether a drug's perturbation signature in the target cell type propagates into off-target effects in cell types like the liver or heart. This is tractable because the failure mode is mechanistic and learnable: drugs that cause liver toxicity do so through identifiable transcriptional signatures that precede cell death. Models trained on these signatures can flag liability before any animal or human study is run.
The second application is combinatorial perturbation prediction. Because the model learns shared regulatory logic rather than memorizing individual experiments, it can, in principle, predict the effects of combinations of perturbations that were never observed during training. This matters because most diseases are not caused by a single molecular failure. Hitting two targets simultaneously, or combining a drug with a genetic intervention, opens a combinatorial space that is entirely intractable to explore experimentally.
A model that understands how individual pathways behave and interact can predict the joint effects of hitting multiple nodes simultaneously. This also opens the door to drug repurposing: identifying combinations of existing approved drugs that could treat diseases for which they were never developed. Thalidomide, a sedative that caused widespread birth defects and was pulled from the market in the 1960s, was later found to drive remission in multiple myeloma, a blood cancer that had seen almost no treatment progress in decades. It became the first drug in a generation to meaningfully treat that cancer and launched an entire new class of medicines. A virtual cell could systematically search for similar surprises across the full space of approved drugs.
The third application is inverse perturbation prediction, and it is arguably the most powerful. Rather than asking what happens to a cell when a drug is applied, it asks the reverse: given a diseased cell state and a desired healthy state, what perturbation would drive the transition? This reframes drug discovery as a control problem. The disease state and the healthy state are both defined by their gene expression profiles. The model searches the space of possible interventions for the minimal set of perturbations, whether genetic, pharmacological, or both, that would move the cell from one to the other. This is the computational equivalent of finding the minimal intervention needed to move a ball from a disease attractor state “valley” over the hill and into a healthy state.
Clinical Applications
The fourth and most immediate application of virtual cells, which can be used today, is patient stratification in clinical trials.
What patient stratification means is that two patients with the same diagnosis frequently have different diseases at the molecular level. Lupus, inflammatory bowel disease, and rheumatoid arthritis are textbook examples. The response rates to drugs treating these diseases commonly fall between 40 and 60%, and clinicians have no reliable way to predict in advance who will respond between patients, despite the fact that all of them have the disease.
The substructure of how such diseases vary in different patients is visible in the cells themselves, but identifying it requires a model that has learned cell identity. Given a patient's gene expression profile, whether from a tumor biopsy, a blood draw, or a tissue sample, a virtual cell projects that sample into its learned cell-state space and identifies the cells it most closely resembles. The result is a quantitative molecular phenotype that sorts patients who share a single diagnosis into molecularly distinct subgroups, each with its own likely response profile to existing therapies. Pharmaceutical companies could use such virtual cell models to predict how different patients in a future trial will respond to the drug. This is achievable today using data already being collected in routine clinical workflows.
Stratification today works by classification: the model recognizes which subgroup a patient falls into based on similarity to historical data. Once a virtual cell can reliably simulate how a candidate therapy would act on a given cell state, the same patient representation could be used to figure out which intervention, applied to this patient's cells, would most likely move them from their diseased state toward a healthy one. This is inverse perturbation prediction applied at the resolution of a single patient, and it is the bridge from population-level stratification, which sorts patients into groups, to personalized medicine, which selects the optimal therapy for each individual based on the molecular profile of their own cells.
The fifth application extends beyond the individual cell. Because virtual cells learn the rules of cellular behavior, they serve as natural building blocks for higher-order models. By mapping virtual cells onto spatial and tissue-resolved data and learning how cells interact with each other and with their environment, it becomes possible to simulate perturbation effects at the tissue, organ, and eventually patient level. A virtual tissue built from virtual cells could capture systems-level effects that are entirely invisible to models focused on a single cell type in isolation, and could eventually form the basis for patient-specific simulations that predict how an individual will respond to a treatment based on the molecular profile of their cells.
Where Virtual Cells Work Best
Virtual cells are not a universal tool. They are most useful for a specific class of problems: diseases in which the pathology is encoded in the regulatory state of identifiable cell populations, and in which therapy amounts to deliberately shifting those states. Three conditions define this class.
The first is that the disease can be compressed to the cellular level. There must be specific cell types whose states are causally linked to the disease. This is true for most cancers, autoimmune diseases, fibrotic diseases, and neurodegeneration. It is less clearly true for diseases driven primarily by tissue mechanics, blood flow, or structural failure, such as bone fractures, aneurysms, or heart valve defects. These are problems of physics, not cellular programming, and fall outside the scope of what a virtual cell can address.
The second condition concerns the structure of how the disease operates at the cellular level. It helps to distinguish between two types of therapeutically relevant cells: driver cells, whose aberrant state directly causes the pathology, and effector cells, whose state can be leveraged to alter the environment in which the driver cells live. For example, myofibroblasts in fibrosis and tumor cells in cancer are driver cells. CD8+ T cells in cancer are effector cells; if they can be shifted out of an exhausted state, they can directly kill tumor cells. Virtual cells are useful for reprogramming both. This distinction also raises a practical consideration: shifting a driver cell into a healthy state is not durable if the environmental conditions that caused the original transition remain unchanged. Effective therapy, therefore, requires either a new cell state robust enough to resist those conditions or an intervention that simultaneously remodels the environment.
The third condition is that the disease state is stable and self-sustaining. Virtual cells are most useful when a cell has locked into a pathological configuration through internal regulatory feedback and stays there even after the original trigger is removed. This is what makes many diseases so hard to treat: the drug may suppress symptoms while the patient is taking it, but if the underlying cellular state remains intact, the disease returns when the drug is stopped. Durable treatment requires not just suppressing a disease state but destabilizing it, pushing the cell into a different configuration that is itself self-sustaining. This is precisely what inverse perturbation prediction aims to achieve.
An additional variable is regulatory complexity. Not every stable disease state requires a virtual cell model. Some diseases are driven by a single dominant molecular driver, and conventional drug discovery handles these well. Chronic myeloid leukemia is the clearest example, in which a genetic rearrangement creates a permanently activated protein called BCR-ABL that drives multiple cancer-promoting pathways simultaneously. The drug imatinib, which targets BCR-ABL directly, is one of the most successful drugs in oncology history precisely because the complexity of the disease funneled through a single bottleneck. Virtual cells become necessary when the disease state arises from the coordinated activity of hundreds or thousands of interacting regulators rather than a single dominant one. In these cases, no amount of pathway analysis or biological intuition can reliably identify the right intervention. Computational search across the full regulatory network is the only way in.
Diseases Suited to Virtual Cell Models
When all three conditions are met, the same computational problem recurs across disease areas: a cell trapped in the wrong state, held there by a regulatory network too complex to reason about from known biology alone. What follows is a survey of the disease classes where this structure appears most clearly.
Fibrosis
Fibrosis is a common path to organ failure across virtually every major organ system, and no approved therapy can reverse it. In the lung, liver, kidney, and heart, the underlying cellular process is similar: activated cells called myofibroblasts deposit excessive scar tissue and progressively replace functional tissue.
The two available antifibrotic drugs slow decline in lung function but cannot stop it, because the myofibroblast is locked into a self-sustaining state maintained by multiple feedback loops. The surrounding scar tissue is stiff, and that stiffness signals back to keep the myofibroblast activated. The cell also secretes a molecule called TGF-beta that reinforces its own gene expression. Chemical modifications to the cell's DNA lock the whole program in place.
Each loop independently sustains the disease, so blocking any one leaves the others intact. And yet the state is reversible: studies have shown that during fibrosis regression, roughly half of myofibroblasts revert to a quiescent state, establishing that the attractor can be destabilized. The challenge is identifying which combination of interventions will do so deliberately, across a regulatory network too complex to reason about from known biology alone.
Neurodegeneration
Over 55 million people live with neurodegenerative diseases like Alzheimer's and Parkinson's. The neurons that die in these diseases are generally not reprogrammable; they are gone. But the brain's resident immune cells, called microglia, modulate whether and how fast neurodegeneration occurs, and they represent a therapeutic opportunity.
Single-cell sequencing has revealed that in Alzheimer's disease, a subset of microglia adopts a distinct transcriptional program called disease-associated microglia, or DAM, in which debris-clearing and fat-processing genes are switched on while normal housekeeping genes are switched off. This same signature appears across mouse models of Alzheimer's, ALS, and multiple sclerosis.
Genetic evidence supports its importance: a variant in the TREM2 gene that disrupts full DAM activation is among the strongest known genetic risk factors for Alzheimer's. The goal is not simply to eliminate this state but to understand which features of it are protective and which are harmful, and to steer microglia toward an optimal configuration. This is precisely the kind of high-dimensional regulatory problem that virtual cells are designed to address.
Cancer
Cancer is the second leading cause of death worldwide as of 2026, and drug resistance is responsible for up to 90% of cancer-related mortality. Targeted therapies have transformed outcomes in some cancers, but in most cases, resistance eventually emerges. A growing body of evidence points to a specific mechanism: drug-tolerant persister cells, a small subpopulation that survives treatment not through new mutations but through reversible changes in gene expression and epigenetic state. Crucially, the persister state is reversible: when the drug is removed, these cells can resume normal growth and regain drug sensitivity, confirming that resistance in this case is a stable but not permanent cell state.
This reversibility is also what makes it dangerous: persister cells act as a reservoir from which permanent genetic resistance can eventually evolve. In the best-studied example, melanoma cells treated with targeted therapy switch into a slow-growing, drug-tolerant configuration within a median of nine months. Adding a second targeted drug extends this window only modestly, because the underlying state-switching mechanism is not addressed by either drug. The question is which combinations of perturbations will destabilize the persister state before permanent resistance emerges, a search across a combinatorial space that sequential clinical trial design cannot effectively explore.
Beyond drug resistance, virtual cells may help solve the problem of predicting which patients will respond to treatment in the first place. The most widely used biomarker for checkpoint inhibitor eligibility, PD-L1 expression, predicts response in only 28.9% of FDA-approved indications. The reason is that response depends not on any single marker but on the full cellular ecosystem of the tumor. Early work has shown that virtual cell embeddings can separate checkpoint inhibitor responders from non-responders within populations that standard biomarkers classify as equivalent, suggesting that molecular portraits of the tumor microenvironment could eventually replace crude single-marker cutoffs in trial design.
Autoimmune and Chronic Inflammatory Diseases
An estimated 15 to 25 million Americans suffered from autoimmune diseases as of 2024, including rheumatoid arthritis, inflammatory bowel disease, and lupus, conditions where the immune system attacks the body's own tissues. Current therapies, including anti-TNF antibodies and JAK inhibitors, can be remarkably effective at reducing symptoms, but they do not fix the underlying problem.
A 2019 meta-analysis of roughly 1.3K rheumatoid arthritis patients found that about half of patients relapse when anti-TNF therapy is stopped, even after achieving full remission. The disease was never resolved. It was held in check. This is because the immune cells driving the disease are stuck in a self-reinforcing inflammatory loop, and suppressing their output does not destabilize the loop itself. The goal for virtual cells is not to identify better suppressors but to find the specific combination of perturbations that shifts these cells from their pathological configuration back into a tolerant one, fully resolving the underlying pathology instead of just chronically suppressing it.
Aging and Cellular Senescence
As cells age, some enter a state called senescence: they stop dividing permanently but refuse to die. Each senescent cell secretes hundreds of inflammatory molecules and tissue-degrading enzymes, collectively known as the senescence-associated secretory phenotype, or SASP, which inflame surrounding tissue and push neighboring healthy cells into senescence. The immune system normally clears these cells, but this capacity erodes with age, creating a self-reinforcing cycle of accumulation and deterioration. Senescent cell accumulation drives osteoarthritis, pulmonary fibrosis, atherosclerosis, Alzheimer's disease, and type 2 diabetes.
Drugs that selectively kill senescent cells have entered clinical trials, but the field has hit a wall: Unity Biotechnology's lead candidate failed Phase 2 for osteoarthritis, and 30% to 70% of senescent cells survive current treatments. The root cause is heterogeneity. A senescent immune cell in an atherosclerotic plaque and a senescent cartilage cell in an arthritic knee are molecularly distinct states with different vulnerabilities. There is no universal senescent cell marker, and no single-target therapy can address them all. A virtual cell could learn the regulatory architecture of each senescent subtype and predict the minimal perturbation set to destabilize it.
Stem Cell Reprogramming
Hundreds of diseases are caused by the irreversible loss of specific cell types. The 9.5 million people with Type 1 diabetes have lost their insulin-producing beta cells. The 10 million people with Parkinson's disease are losing their dopamine-producing neurons. The premise of stem cell therapy is to manufacture these missing cells.
In 2006, Shinya Yamanaka showed that four transcription factors could revert an adult cell back to a stem cell-like state, creating induced pluripotent stem cells, or iPSCs. We have also been able to convert cells directly from one mature cell type to another. Early clinical results are striking: Vertex's stem cell-derived islet therapy restored insulin production in all 12 patients dosed, with 10 of 12 achieving insulin independence at one year.
But each of these successes required years of empirical protocol optimization, because the regulatory transitions involved are governed by dozens of interacting signals, and less than 1% of cells successfully reprogram under standard conditions. Protocol development today is brute-force screening, testing combinations of growth factors and small molecules over months or years for each target cell type. A virtual cell model that captures the full regulatory landscape could predict which perturbations, applied in which sequence and at which dose, would most efficiently drive cells through the desired transition, turning years of empirical trial and error into a tractable computational problem.
Common Structures Across Indications
Across every disease examined here, the therapeutic problem has the same structure. In fibrosis, a myofibroblast is locked into a scarring program. In neurodegeneration, microglia are stuck in a configuration that may accelerate rather than slow neuronal loss. In cancer, persister cells occupy a drug-tolerant state that seeds permanent resistance, while exhausted T cells have lost their ability to kill cancer cells. In autoimmune disease, immune cells are trapped in a self-reinforcing inflammatory loop. In senescence, aged cells refuse to die and poison their neighbors. In stem cell therapy, the goal is to push a cell through a precise sequence of states to become something it is not.
These appear to be six distinct problems spanning six fields of medicine, but they are computationally a single problem: given a cell in state A, identify the minimal set of perturbations that will move it to state B. The reason each remains unsolved is not that we lack drugs, transcription factors, or small molecules. It is that the regulatory networks governing cell state are too high-dimensional for any human or simple model to search effectively. Every disease described above has resisted single-target approaches for the same reason: the pathological state is maintained by multiple interlocking feedback loops, and blocking one leaves the others intact.
A virtual cell model encodes what is needed to break this impasse. It represents the full regulatory state of a cell, models how that state changes under perturbation, and searches a combinatorial space that no amount of intuition or sequential experimentation can cover. The same model architecture that predicts how to collapse a fibrotic attractor can also predict how to steer a T cell out of exhaustion or to guide a stem cell through differentiation. It is a single computational framework tailored to a single class of problems that underlie some of the largest unmet needs in medicine.
The diseases that fall outside this framework are equally instructive. Acute infections that resolve with pathogen clearance, traumatic injuries dominated by mechanical tissue disruption, and diseases driven by rare stochastic molecular events rather than stable cellular programs do not satisfy the three criteria and should not be forced into the virtual cell paradigm. Recognizing these boundaries is essential to its rigorous application.
Mapping the Virtual Cell Landscape
The virtual cell ecosystem is rapidly growing and evolving. This section outlines the major nonprofit research institutions and biopharma companies that are leading research and innovation in the virtual cell field.
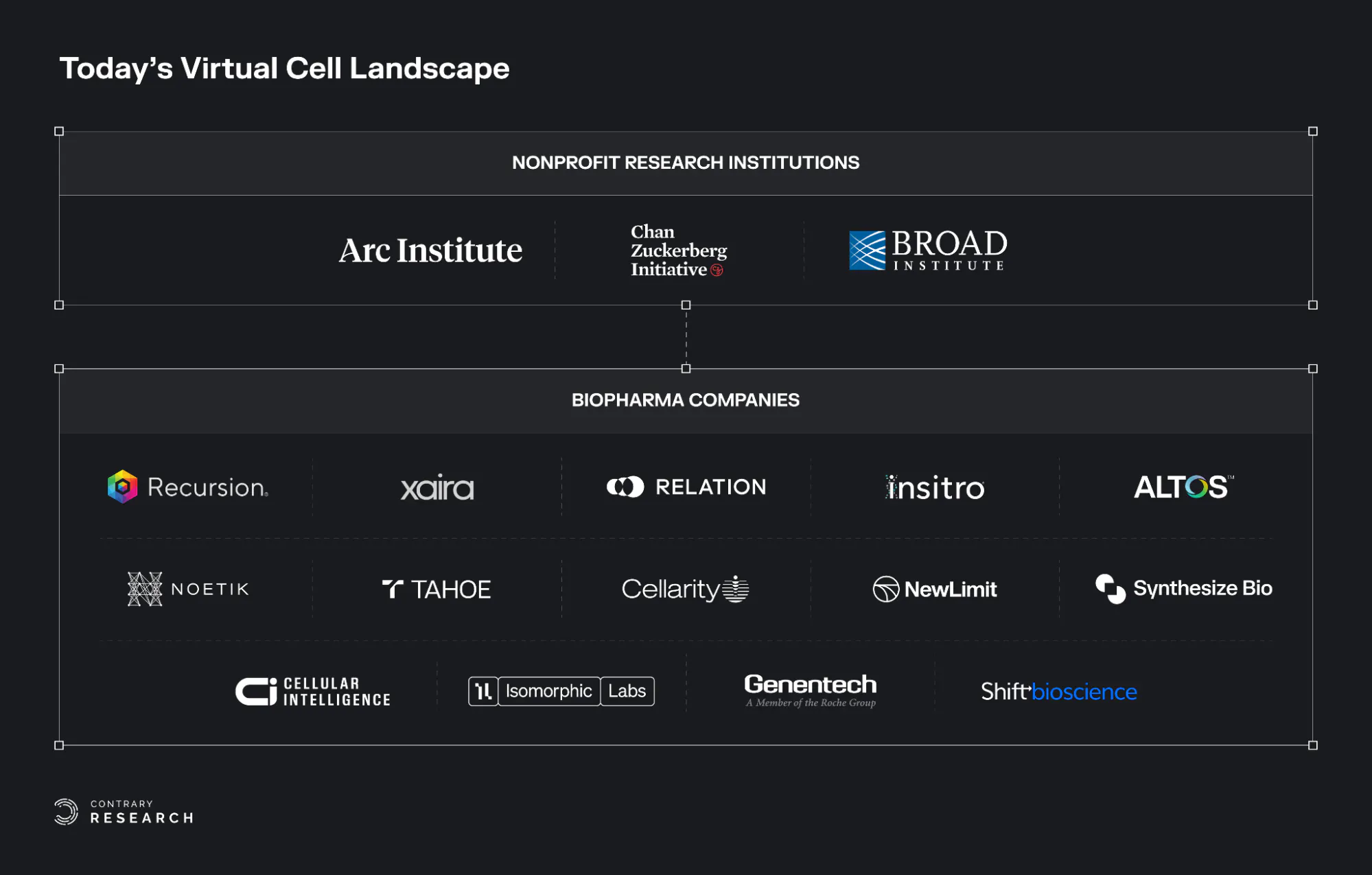
Source: Contrary Research
Nonprofit Research Institutions
Arc Institute: The Arc Institute, co-founded in 2021 with over $650 million in donor funding, has built the Arc Virtual Cell Atlas, an open data resource of over 600 million cells. In June 2025, Arc released State, its first virtual cell model, trained on 170 million observational and over 100 million perturbational cells across 70 cell lines. Arc also launched the Virtual Cell Challenge, an annual open benchmark framed as a Turing test for virtual cells, whose inaugural 2025 edition drew over 5K registrants across 114 countries.
Chan Zuckerberg Initiative: Founded by Priscilla Chan and Mark Zuckerberg in 2015, the Chan Zuckerberg Initiative's CELLxGENE platform is the largest standardized open-source corpus of curated single-cell data and serves as a central data hub for the field. In February 2025, CZI launched the Billion Cells Project to generate one billion cells specifically for virtual cell training. In September 2025, CZI released TranscriptFormer, the first multi-species generative model for single-cell transcriptomics, trained on 112 million cells from 12 species spanning 1.5 billion years of evolution.
Broad Institute of MIT and Harvard: Founded in 1982, the Broad Institute has focused on interpretability and benchmarking infrastructure rather than scaling a single model. Key outputs include Morph, a framework that predicts single-cell responses to genetic perturbations across cell types and data modalities while using attention mechanisms to infer gene regulatory networks.
Biopharma Companies
Recursion Pharmaceuticals: Founded in 2013, Recursion Pharmaceuticals operates one of the largest high-content cellular imaging platforms in drug discovery, with over 50 petabytes of proprietary biological and chemical data. Its ML research arm published TxPert in 2025, a model for predicting transcriptomic responses to unseen perturbations, and has extended this work to generating in silico predictions of imaging-based cellular assays.
Xaira Therapeutics: Founded in 2023, Xaira Therapeutics released X-Cell in March 2026, a 4.9 billion parameter model trained on the largest genome-wide Perturb-seq dataset reported to date, covering 25.6 million perturbed single-cell transcriptomes across 16 experimental contexts. The company reports zero-shot generalization to unseen cell types and claims perturbation prediction follows power-law scaling when model size and biological diversity increase together.
Relation Therapeutics: Founded in 2019, Relation Therapeutics generates single-cell multi-omic perturbation data directly from patient tissue rather than cell lines or iPSCs, producing what it calls functional cell atlases of disease states in human tissue. This approach has attracted a $45 million contract with GSK and a $55 million contract with Novartis.
Insitro: Founded in 2018, Insitro has made a distinctive bet that how a cell looks can reveal as much about its state as what genes it expresses. Its CellPaint-POSH platform combines pooled CRISPR screening with Cell Painting imaging and self-supervised Vision Transformers to map gene function at scale without predefined biomarkers. Across a screen of 1.6K genes, the platform reconstructed known biological networks purely from morphological features, with self-supervised learning outperforming classical expert-engineered image analysis. The company has raised a total of $643 million in funding and maintains a partnership with Bristol Myers Squibb focused on ALS.
Altos Labs: Altos Labs was founded in 2021. Focused on rejuvenation biology and aging interventions, Altos Labs’s most visible contribution to the virtual cell ecosystem is PerturBench, an open-source benchmarking framework published at NeurIPS 2025 that standardizes evaluation of perturbation-prediction models across diverse datasets, tasks, and metrics. Altos also won the Generalist Prize at the Arc Institute's inaugural Virtual Cell Challenge at NeurIPS 2025, using a flow-matching generative model pre-trained on roughly seven million cells from public and proprietary perturbation screens.
Noetik: Noetik, founded in 2022, takes a fundamentally different approach by modeling cells within their native tissue context rather than in isolation. Its spatial virtual cell model, OCTO-VC, is trained on nearly 40 million spatially resolved cells from over 1K patient tumor biopsies. The model simulates gene expression of virtual cells placed in real tissue microenvironments, for example, predicting whether a T cell dropped into a specific region of a tumor would be activated or suppressed. In January 2026, Noetik signed a five-year licensing deal with GSK worth $50 million in upfront capital for access to OCTO-VC in lung and colorectal cancer.
Tahoe Therapeutics: Founded in 2022, Tahoe Therapeutics released the Tahoe-100M atlas in February 2025, containing 100 million single-cell profiles across 1.1K small-molecule drug treatments in 50 cancer cell lines, the largest single-cell perturbation dataset to date, with over 250K downloads. In January 2026, Tahoe announced a partnership with Arc Institute and CZ Biohub to generate over 120 million additional data points across 225K drug-patient interactions.
Cellarity: Cellarity, founded by Flagship Pioneering in 2017, was among the earliest companies to frame drug discovery as a cell state problem. Its lead drug, CLY-124, discovered entirely through its platform, is a first-in-class oral therapy for sickle cell disease that reactivates fetal hemoglobin through a previously unexplored target without the cytotoxicity of existing treatments.
NewLimit: NewLimit, founded in 2022, applies the virtual cell thesis directly to aging, using machine learning to search the combinatorial space of transcription factor combinations that can reprogram old cells toward youthful function. The company claims to have tested over 1K times more reprogramming payloads than the rest of the field combined, discovering 16 payloads that treat disease in animal models, and has selected its first preclinical candidate for alcohol-related liver disease. NewLimit is backed by $247.2 million in total funding, including an investment from Eli Lilly, with plans to enter the clinic in the next few years.
Synthesize Bio: Synthesize Bio, founded in 2023, takes a different approach to virtual cells by generating synthetic gene expression data rather than predicting perturbation responses. Its foundation model GEM-1 generates bulk and single-cell RNA-seq profiles from natural-language experimental descriptions, trained on what the company calls the largest curated RNA-seq corpus ever assembled. The primary use case is augmenting small clinical trial cohorts with synthetic patients to de-risk late-stage decisions.
Isomorphic Labs: A DeepMind spinout founded in 2012 and led by DeepMind founder Demis Hassabis, Isomorphic has articulated a roadmap progressing from proteins to pathways to cells. Its current focus is molecular-scale: in February 2026, it released IsoDDE, a drug design engine that more than doubles AlphaFold 3's accuracy on generalization benchmarks and predicts binding affinities faster and more cheaply than physics-based methods. The company has raised a total of $601.7 million in funding. Teams are reportedly assembling components of a "virtual cell" internally to simulate how biological networks respond to drug perturbations, but no cell-scale model has been publicly released. The virtual cell layer remains a stated ambition rather than a shipped product.
Genentech: Founded in 1976, its R&D is led by Aviv Regev, a pioneer of single-cell genomics and co-founder of the Human Cell Atlas. Genentech likely operates the most ambitious internal virtual cell program in pharmaceutical R&D. Publicly released tools include SCimilarity, a metric-learning framework published in Nature that enables rapid queries across a 23.4-million-cell atlas, and Presage, a perturbation response prediction framework. Unlike startups that must publish to attract partners, much of Genentech's virtual cell work remains internal.
Shift Bioscience: Founded in 2017 at the Gurdon Institute, Shift uses virtual cell models and proprietary single-cell aging clocks to discover genes that reverse cellular age without altering cell identity. The platform has identified over 190 such genes, leading to SB000, a single target that reverses DNA methylation clocks across multiple cell types, now progressing toward therapeutics for age-driven hearing loss, MASH, and systemic sclerosis. Shift also published research showing that reports of poor virtual cell model performance largely reflect metric miscalibration, with models consistently outperforming baselines on well-calibrated metrics across 14 Perturb-seq datasets.
The Next Phase of Virtual Cells
Every disease examined in this essay, including fibrosis, neurodegeneration, cancer, autoimmune disease, and aging, shares the same computational structure: a cell trapped in the wrong state, held there by a regulatory network too complex for any human to search by intuition alone. For decades, we have thrown single-target drugs at these networks and watched them rewire around the drug's effect. This has culminated in $2 billion and 10 years spent per drug and a 90% failure rate as the cost of doing business.
Virtual cells do not yet solve this problem. No model today can reliably predict how an arbitrary drug will behave in an arbitrary patient. The gap between genetic perturbation prediction in cell lines and pharmacological response prediction in human tissue remains real. The field is young, the benchmarks are still being written, and the hardest generalization tests are still ahead.
But for the first time, the necessary conditions are converging for this to change. Sequencing costs have collapsed to the point where perturbation datasets of hundreds of millions of cells exist. AI architectures have demonstrated that biological rules can be learned from data without being programmed by hand. Regulatory frameworks are shifting to accept computational evidence alongside, and eventually in place of, animal data. And the first models are beginning to outperform baselines on held-out perturbations that would have been unthinkable five years ago.
The drug discovery industry has spent the last four decades watching Eroom's Law compound. Every year, drugs get more expensive to develop and are no more likely to succeed. Virtual cells represent a credible mechanism to reverse that trend: not by making the same process cheaper, but by replacing the process itself with one built on human data, computational search, and AI models that improve with scale.
The question is no longer whether it is possible to build a computational model of cellular behavior. The question is whether we will do it fast enough. Thirty million people are living with Alzheimer's. Ten million new cancer diagnoses are made every year. Hundreds of millions suffer from autoimmune and fibrotic diseases with no curative therapy. These patients cannot wait for perfect models. They need better ones, built now, improved continuously, and held to rigorous standards.
